02_Simulated_datasets¶
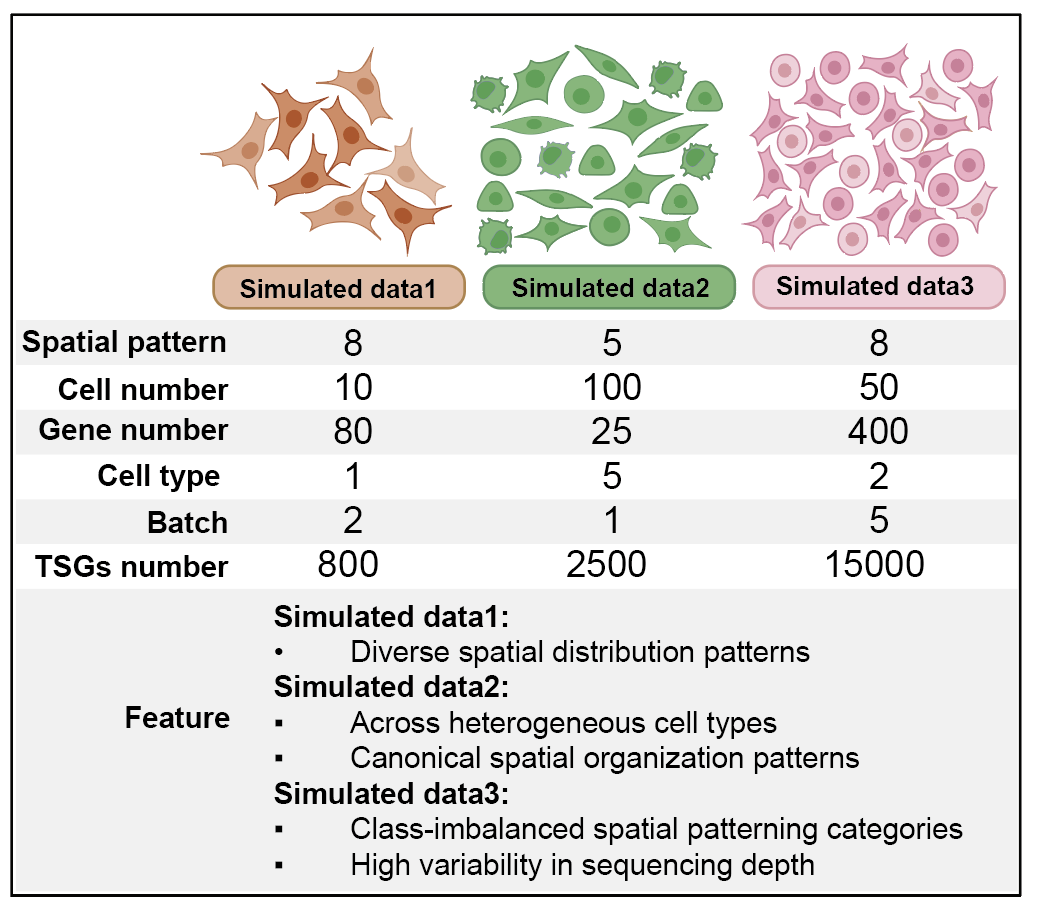
1_dataset details¶
| Dataset | Cell number | Gene number | Cell type | TSGs | Pattern | Ratio | Batch |
|---|---|---|---|---|---|---|---|
| Simulated_data1 | 10 | 80 | 1 | 800 | 8 | 5 | 2 |
| Simulated_data2 | 100 | 25 | 5 | 2500 | 5 | 5 | 1 |
| Simulated_data3 | 50 | 400 | 2 | 15000 | 8 | 10 | 5 |
Tips¶
Using the built-in 318 cell templates from the simfish python package, 10/20 cells were randomly selected;- Generated RNA spatial localization data for 8 gene spatial distribution patterns: Random, Foci, Intranuclear, Extranuclear, Perinuclear, Pericellular, Protrusion, and Radial (ours);
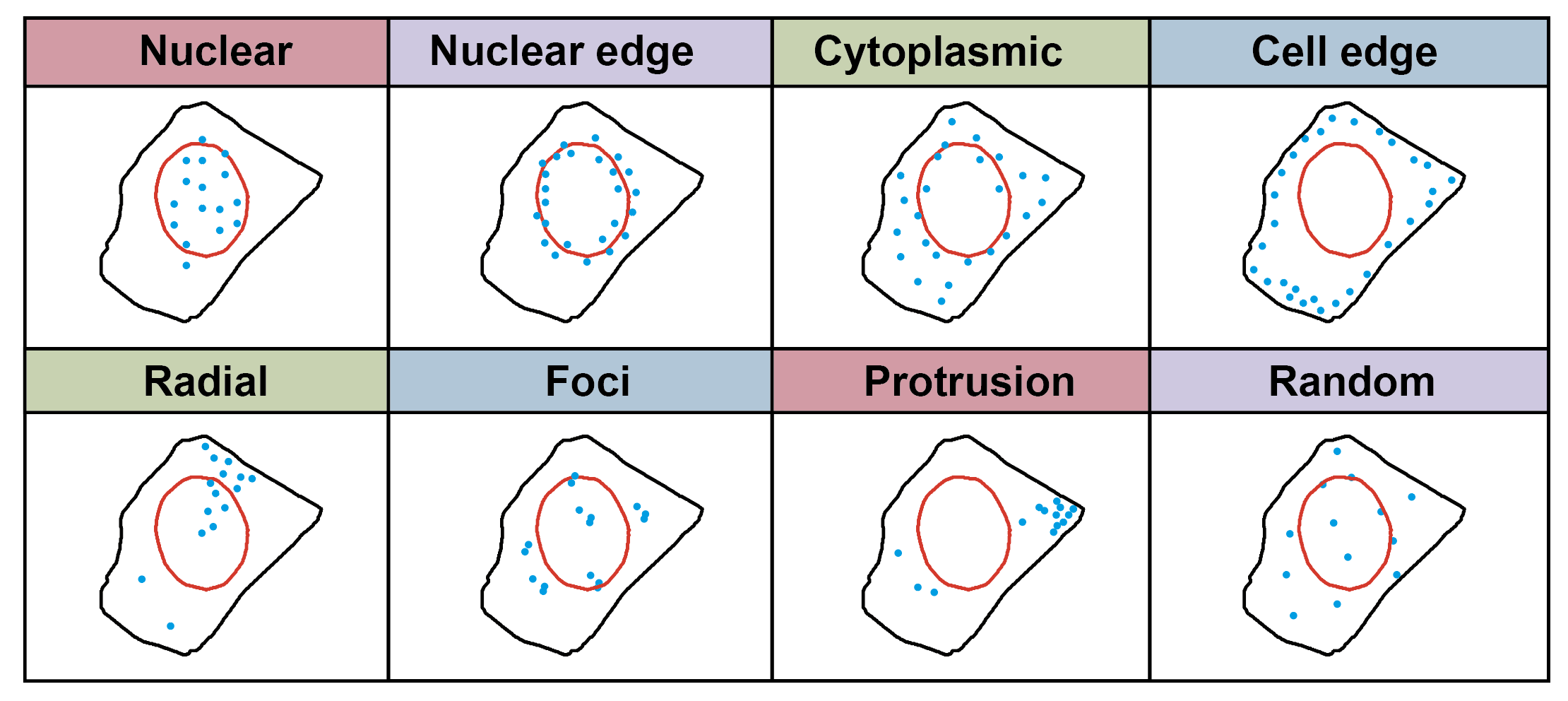
Each spatial pattern simulated 5 different intensities of spatial distribution: 60%, 70%, 80%, 90%, and 100%, with higher intensity leading to more pronounced spatial distribution patterns;
Each spatial pattern also simulated 25, 50, 100, 150, and 200 different transcript numbers;
Different expression intensities and different transcript numbers of the same spatial pattern can be viewed as a gene expressing that pattern;
Foci is a special pattern that does not require specification of transcript numbers.
2_GRASP preprocessing¶
step1: Load data¶
dataset = "simulated1"
outfile = f'../1_input/pkl_data/{dataset}_data_dict.pkl'
with open(outfile, 'rb') as f:
pickle_dict = pickle.load(f)
df_registered = pickle_dict['df_registered']
cell_radii = pickle_dict['cell_radii']
cell_boundary = pickle_dict['cell_boundary']
nuclear_boundary = pickle_dict['nuclear_boundary']
nuclear_boundary_df_registered = pickle_dict['nuclear_boundary_df_registered']
step2: Visualize the original and normalized TSGs¶
path = "../2_scaled_cell"
cep.plot_raw_gene_distribution(dataset, cell_boundary, nuclear_boundary, df_registered, path)
cep.plot_register_gene_distribution(dataset, df_registered, path, nuclear_boundary_df_registered)
Processing cells: 100%|██████████| 10/10 [11:26<00:00, 68.62s/it]
All cell images have been saved to ../2_scaled_cell/simulated1/raw_gene/cell_cell_257
Plotting per cell: 100%|██████████| 10/10 [12:38<00:00, 75.84s/it]
step3: Cell partitioning¶
import utils_code.partition as pat
gene_list = df_registered['gene'].unique()
cell_list = df_registered['cell'].unique()
dir = f"../4_partition_same/{dataset}_partition/"
if not os.path.exists(dir):
os.makedirs(dir)
for n_sectors in range(30, 31, 10):
for m_rings in range(15, 16, 5):
for target_cell in cell_list:
df = df_registered[df_registered['cell'] == target_cell]
genes = df['gene'].unique()
k_neighbor = int((n_sectors * m_rings) / 10)
plot_dir = os.path.join(dir,f"{target_cell}/{target_cell}_{n_sectors}_{m_rings}_k{k_neighbor}")
if not os.path.exists(plot_dir):
os.makedirs(plot_dir)
print(f"This is [target_cell: {target_cell}] - [n_sectors: {n_sectors}] - [m_rings: {m_rings}], and k_neighbor is {k_neighbor}")
nuclear_boundary_df = nuclear_boundary_df_registered[nuclear_boundary_df_registered['cell'] == target_cell]
for gene in tqdm(gene_list, desc="Processing genes"):
df_filtered = df[df['gene'] == gene]
r = 1 # r = cell_radii[target_cell]
count_matrix, center_points, point_counts, is_virtual, is_edge = pat.count_points_in_areas_same(df_filtered, n_sectors, m_rings, r)
nuclear_positions = pat.classify_center_points_with_edge(center_points, nuclear_boundary_df, is_edge)
edges = pat.build_graph_k_nearest(center_points, k=k_neighbor)
G = pat.build_graph_with_networkx(center_points, edges, is_virtual)
pat.save_node_data_to_csv_old(center_points, is_virtual, plot_dir, gene, point_counts, k=k_neighbor, nuclear_positions=nuclear_positions)
pat.plot_cell_partition_heatmap(target_cell, gene, point_counts, n_sectors, m_rings, r, plot_dir, nuclear_boundary_df)
step4: Enhancement of TSGs¶
import utils_code.augumentation as aug
n_sectors = 30
m_rings = 15
k_neighbor = int((n_sectors * m_rings) / 10)
dropout_ratios = [0.1, 0.2, 0.3]
dir = f"../4_partition_same/{dataset}_partition/"
gene_list = df_registered['gene'].unique()
cell_list = df_registered['cell'].unique()
for cell in tqdm(cell_list, desc="Processing all cells", leave=True):
path = f"{dir}/{cell}/{cell}_{n_sectors}_{m_rings}_k{k_neighbor}"
save_path = f"{dir}/{cell}/{cell}_{n_sectors}_{m_rings}_k{k_neighbor}_aug"
if not os.path.exists(save_path):
os.makedirs(save_path)
for gene in tqdm(gene_list, desc="Processing all genes", leave=True):
nodes_file = f'{path}/{gene}_node_matrix.csv'
adj_file = f'{path}/{gene}_adj_matrix.csv'
if not os.path.exists(nodes_file) or not os.path.exists(adj_file):
print(f"Skipping {gene} in {cell} (file not found).")
continue
node_matrix = pd.read_csv(nodes_file)
adj_matrix = pd.read_csv(adj_file)
random_angle = random.uniform(0, 360)
node_matrix_rotated = aug.rotate_nodes(node_matrix.copy(), random_angle)
real_nodes_count = (node_matrix_rotated['is_virtual'] == 0).sum()
if real_nodes_count >= 10:
if real_nodes_count <= 100:
dropout_ratio = dropout_ratios[0]
elif real_nodes_count > 100 and real_nodes_count <= 150:
dropout_ratio = dropout_ratios[1]
else:
dropout_ratio = dropout_ratios[2]
# print(f"The gene {gene} needs to drop out {dropout_ratio}.")
adj_matrix_dropped, node_matrix_dropped = aug.dropout_nodes(adj_matrix.copy(), node_matrix_rotated.copy(), dropout_ratio)
adj_matrix_dropped.to_csv(f"{save_path}/{gene}_adj_matrix.csv", index=False)
node_matrix_dropped.to_csv(f"{save_path}/{gene}_node_matrix.csv", index=False)
# aug.plot_graph(adj_matrix, node_matrix,adj_matrix_dropped, node_matrix_dropped, f"{cell}_{gene}", save_path)
else:
# print(f"The gene {gene} not need to drop out.")
adj_matrix.to_csv(f"{save_path}/{gene}_adj_matrix.csv", index=False)
node_matrix_rotated.to_csv(f"{save_path}/{gene}_node_matrix.csv", index=False)
# aug.plot_graph(adj_matrix, node_matrix, adj_matrix, node_matrix_rotated, f"{cell}_{gene}_original", save_path)
3_GRASP training¶
step5: Load all TSGs to prepare for training¶
cell_list = df_registered['cell'].unique()
gene_list = df_registered['gene'].unique()
cell_numbers = len(cell_list)
gene_numbers = len(gene_list)
n_sectors = 30
m_rings = 15
k_neighbor = int((n_sectors * m_rings) / 10)
path = f"../4_partition_same/{dataset}_partition"
df = pd.read_csv(f"../1_input/label/{dataset}_label.csv")
original_graphs, augmented_graphs = gra.generate_graph_data_target(dataset, df, path, n_sectors, m_rings, k_neighbor)
print(len(original_graphs))
print(len(augmented_graphs))
gene_labels = [data.gene for data in original_graphs]
cell_labels = [data.cell for data in original_graphs]
Processing Graphs: 100%|██████████| 800/800 [3:22<00:00, 7.49it/s]
graphs_number = len(original_graphs)
print(f"cell_numbers:{cell_numbers} - gene_numbers:{gene_numbers} - graphs_number:{graphs_number}")
save_path = f"../5_graph_data"
if not os.path.exists(save_path):
os.makedirs(save_path)
graph_data = {"original_graphs": original_graphs,
"augmented_graphs": augmented_graphs,
"gene_labels": gene_labels,
"cell_labels": cell_labels}
save_file = f"{save_path}/{dataset}_cell{cell_numbers}_gene{gene_numbers}_graph{graphs_number}.pkl"
with open(save_file, 'wb') as f:
pickle.dump(graph_data, f)
print(f"Graph data saved to {save_file}")
cell_numbers:10 - gene_numbers:80 - graphs_number:800
Graph data saved to ../5_graph_data/simulated1_cell10_gene80_graph800.pkl
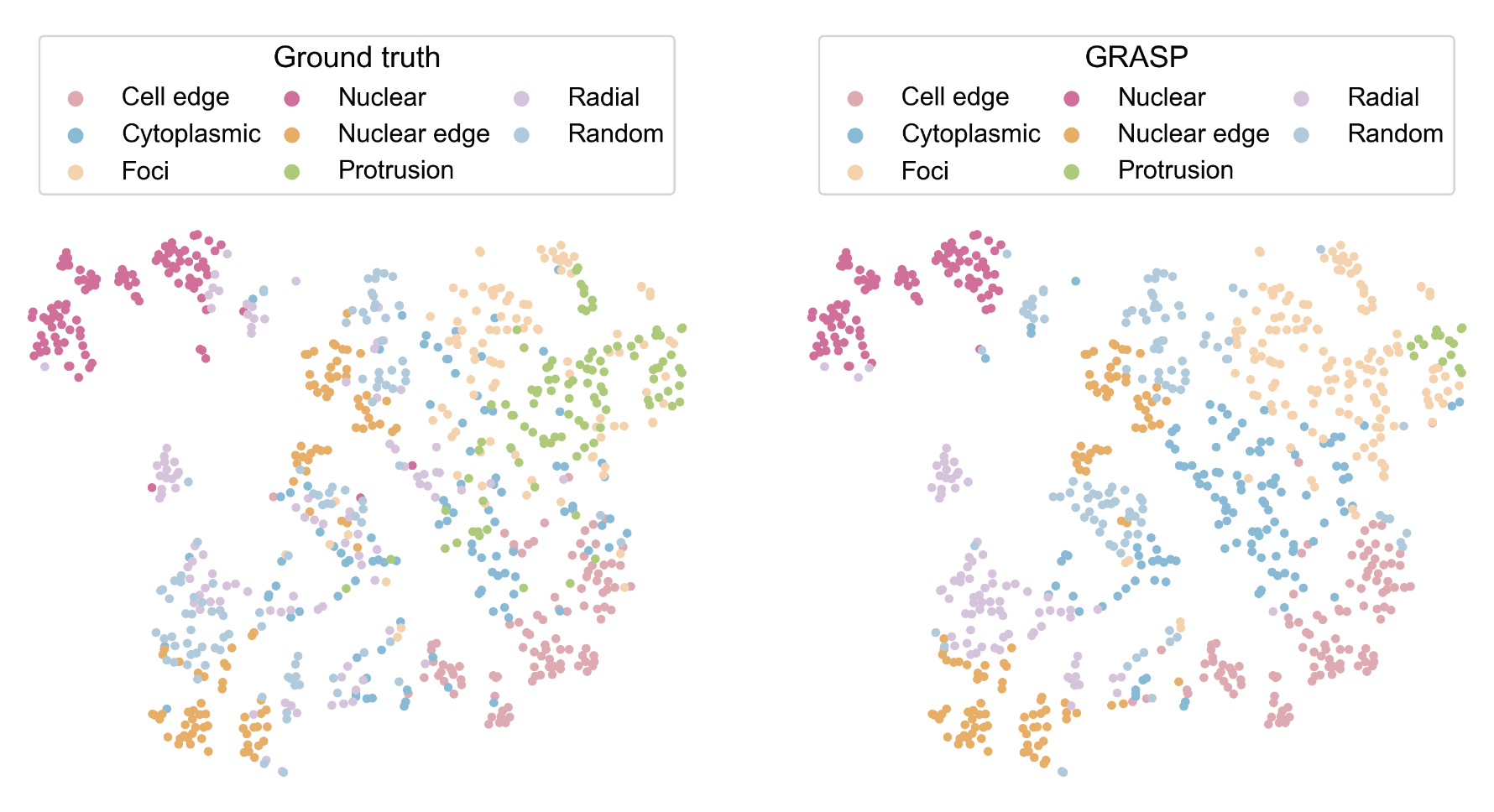
step6: Clustering and identifying spatial localization patterns¶
a, b = 0.4, 0.6
dataset = "simulated1"
path1 = f'../1.5_benchmark/figure/{dataset}/s1_tsne_ours_a{a}_b{b}'
path2 = f'../1.5_benchmark/figure/{dataset}/s1_tsne_gt_a{a}_b{b}'
ours_label = pd.read_csv(f"../1.5_benchmark/method4_ours/{dataset}/ours_label_a{a}_b{b}.csv")
ours_label['pattern'].value_counts()
# 定义 color_map
color_map = {'Nuclear': '#ed6ca4', 'Cytoplasmic': '#7bc4e2', 'Protrusion': '#acd372', 'Nuclear edge':'#fbb05b',
'Cell edge': '#EDABB5', 'Random':'#ACD0E4', 'Foci':'#FFD4AB', 'Radial':'#DDC4E0'}
plot_tsne_by_label(df=ours_label,label_col='pattern',color_map=color_map, title='', save_prefix=path1,
legend_title='GRASP', legend_loc='upper left', legend_bbox=(0.5, 1), legend_ncol=3)
plot_tsne_by_label(df=ours_label, label_col='groundtruth', color_map=color_map, title='', save_prefix=path2,
legend_title='Ground truth', legend_loc='upper left', legend_bbox=(0.5, 1.0), legend_ncol=3)