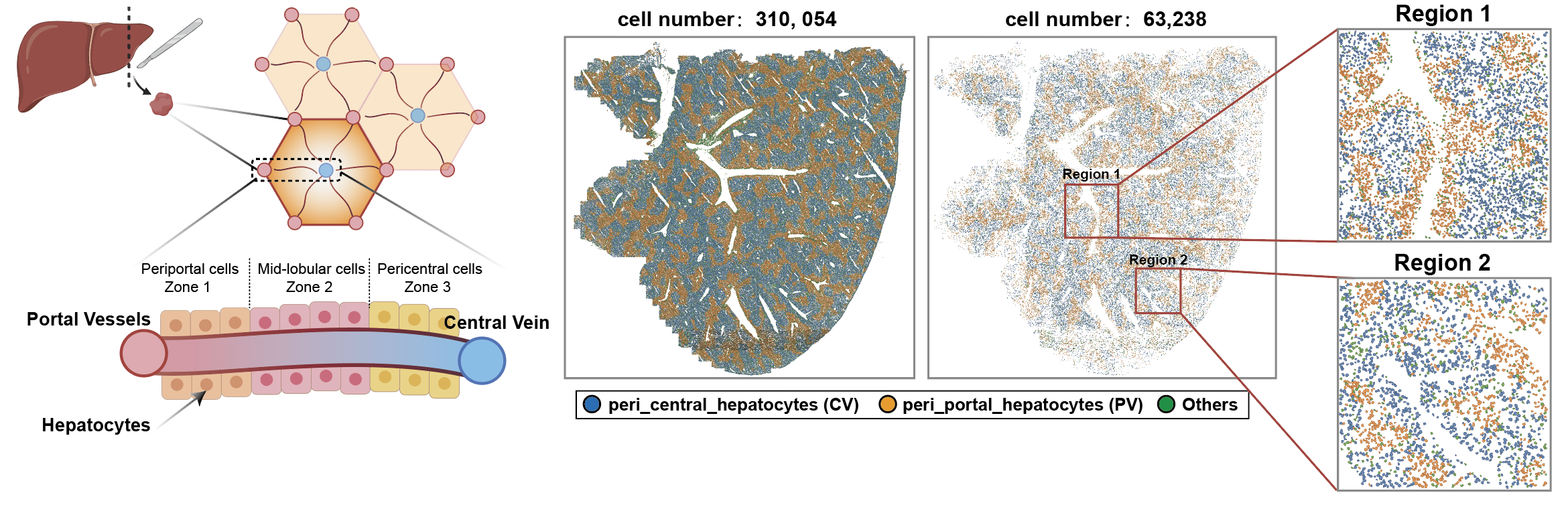
06_MERFISH_liver¶
1_dataset details¶
| Dataset | Cell number | Gene number | Graph | Pattern |
|---|---|---|---|---|
| region1_CV | 1711 | 136 | 86074 | 5 |
| region1_PV | 1708 | 143 | 79172 | 5 |
| region2_CV | 936 | 126 | 44910 | 5 |
| region2_PV | 747 | 124 | 33523 | 5 |
How do you divide cells and assign transcripts?¶
Cell and nuclear boundaries were delineated on the central optical plane (Z = 6 µm) using Vizgen’s post-processing tool (vpt 1.3.0) and Cellpose (1.0.2). Whole-cell masks were generated with the “cyto2” model for both DAPI and Poly T channels, while nuclei were segmented using the “nuclei” model on DAPI alone. Masks were projected into a unified coordinate system, then simplified, smoothed, and filtered to exclude objects < 500 px². Cells were paired with nuclei based on ≥ 50% overlap; unmatched nuclei or cells were removed, and excess overlapping nuclei were excluded. These matched masks were used to assign MERSCOPE-detected RNA molecules.
2_GRASP preprocessing¶
step1: Load data¶
dataset = "merscope_liver_data_region1_portal" # merscope_liver_data_region1_central
outfile = f'../1_input/pkl_data/{dataset}_data_dict.pkl'
with open(outfile, 'rb') as f:
pickle_dict = pd.read_pickle(f)
df_registered = pickle_dict['df_registered']
cell_radii = pickle_dict['cell_radii']
cell_boundary = pickle_dict['cell_boundary']
nuclear_boundary = pickle_dict['nuclear_boundary']
nuclear_boundary_df_registered = pickle_dict['nuclear_boundary_df_registered']
step2: Cell partitioning¶
import utils_code.partition as pat
from multiprocessing import Pool, cpu_count
dataset = "merscope_liver_data_region1_portal"
dir = f"../4_partition_same/{dataset}_partition/"
os.makedirs(dir, exist_ok=True)
n_sectors = 30
m_rings = 15
k_neighbor = int((n_sectors * m_rings) / 10)
r = 1
result = pd.read_csv(f"../3_filter/{dataset}/load_graph_data.csv")
print("Number of TSGs:", result.shape)
df_registered_group = None
nuclear_boundary_group = None
def init_globals(df_reg, nuclear_boundary_reg):
global df_registered_group, nuclear_boundary_group
df_registered_group = df_reg.groupby("cell")
nuclear_boundary_group = nuclear_boundary_reg.groupby("cell")
def process_row(row):
target_cell = row["cell"]
target_gene = row["gene"]
try:
df = df_registered_group.get_group(target_cell)
df_filtered = df[df["gene"] == target_gene]
if df_filtered.empty:
return
nuclear_boundary_df = nuclear_boundary_group.get_group(target_cell)
except KeyError:
return
plot_dir = os.path.join(dir, f"{target_cell}/{target_cell}_{n_sectors}_{m_rings}_k{k_neighbor}")
csv_path = os.path.join(plot_dir, f"{target_gene}_node.csv")
if os.path.exists(csv_path):
return
os.makedirs(plot_dir, exist_ok=True)
count_matrix, center_points, point_counts, is_virtual, is_edge = pat.count_points_in_areas_same(df_filtered, n_sectors, m_rings, r)
nuclear_positions = pat.classify_center_points_with_edge(center_points, nuclear_boundary_df, is_edge)
edges = pat.build_graph_k_nearest(center_points, k=k_neighbor)
G = pat.build_graph_with_networkx(center_points, edges, is_virtual)
pat.save_node_data_to_csv_old(center_points, is_virtual, plot_dir, target_gene, point_counts, k=k_neighbor, nuclear_positions=nuclear_positions)
# pat.plot_cell_partition_heatmap(target_cell, target_gene, point_counts, n_sectors, m_rings, r, plot_dir, nuclear_boundary_df)
if __name__ == "__main__":
import multiprocessing
with Pool(processes=cpu_count(), initializer=init_globals,
initargs=(df_registered, nuclear_boundary_df_registered)) as pool:
list(tqdm(pool.imap_unordered(process_row, [row for _, row in result.iterrows()]), total=result.shape[0], desc="In parallel processing"))
Number of TSGs: (79172, 2)
In parallel processing: 100%|██████████| 79172/79172 [50:15<00:00, 24.02it/s]
step3: Enhancement of TSGs¶
import utils_code.augumentation as aug
import random
dataset = "merscope_liver_data_region1_portal"
n_sectors = 30
m_rings = 15
k_neighbor = int((n_sectors * m_rings) / 10)
dropout_ratios = [0.1, 0.2, 0.3]
cell_list = df_registered['cell'].unique()
gene_list = df_registered['gene'].unique()
dir = f"../4_partition_same/{dataset}_partition/"
for cell in tqdm(cell_list, desc="Processing all cells", leave=True):
path = f"{dir}/{cell}/{cell}_{n_sectors}_{m_rings}_k{k_neighbor}"
save_path = f"{dir}/{cell}/{cell}_{n_sectors}_{m_rings}_k{k_neighbor}_aug"
if not os.path.exists(save_path):
os.makedirs(save_path)
# for gene in tqdm(gene_list, desc="Processing all genes", leave=True):
for gene in gene_list:
nodes_file = f'{path}/{gene}_node_matrix.csv'
adj_file = f'{path}/{gene}_adj_matrix.csv'
if not os.path.exists(nodes_file) or not os.path.exists(adj_file):
# print(f"Skipping {gene} in {cell} (file not found).")
continue
node_matrix = pd.read_csv(nodes_file)
adj_matrix = pd.read_csv(adj_file)
random_angle = random.uniform(0, 360)
# print(random_angle)
node_matrix_rotated = aug.rotate_nodes(node_matrix.copy(), random_angle)
real_nodes_count = (node_matrix_rotated['is_virtual'] == 0).sum()
if real_nodes_count >= 10:
if real_nodes_count <= 100:
dropout_ratio = dropout_ratios[0]
elif real_nodes_count > 100 and real_nodes_count <= 150:
dropout_ratio = dropout_ratios[1]
else:
dropout_ratio = dropout_ratios[2]
# print(f"The gene {gene} needs to drop out {dropout_ratio}.")
adj_matrix_dropped, node_matrix_dropped = aug.dropout_nodes(adj_matrix.copy(), node_matrix_rotated.copy(), dropout_ratio)
# adj_matrix_add, node_matrix_add = add_nodes(adj_matrix.copy(), node_matrix_rotated.copy(), add_ratio)
adj_matrix_dropped.to_csv(f"{save_path}/{gene}_adj_matrix.csv", index=False)
node_matrix_dropped.to_csv(f"{save_path}/{gene}_node_matrix.csv", index=False)
# aug.plot_graph(adj_matrix, node_matrix,adj_matrix_dropped, node_matrix_dropped, f"{cell}_{gene}", save_path)
else:
# print(f"The gene {gene} not need to drop out.")
adj_matrix.to_csv(f"{save_path}/{gene}_adj_matrix.csv", index=False)
node_matrix_rotated.to_csv(f"{save_path}/{gene}_node_matrix.csv", index=False)
# aug.plot_graph(adj_matrix, node_matrix, adj_matrix, node_matrix_rotated, f"{cell}_{gene}_original", save_path)
3_GRASP training¶
step4: Load all TSGs to prepare for training¶
import pandas as pd
import gnn_model.gcn_cl as gcl
import gnn_model.graphloader as gra
import os
import pickle
dataset = "merscope_liver_data_region1_portal"
n_sectors = 30
m_rings = 15
k_neighbor = int((n_sectors * m_rings) / 10)
df = pd.read_csv(f"../3_filter/{dataset}/load_graph_data.csv")
print(df.shape)
cell_numbers = len(df['cell'].unique())
gene_numbers = len(df['gene'].unique())
print(f"cell_numbers:{cell_numbers} - gene_numbers:{gene_numbers}")
path = f"../4_partition_same/{dataset}_partition"
original_graphs, augmented_graphs = gra.generate_graph_data_target_parallel(dataset, df, path, n_sectors, m_rings, k_neighbor)
gene_labels = [data.gene for data in original_graphs]
cell_labels = [data.cell for data in original_graphs]
graphs_number = len(original_graphs)
save_path = f"../5_graph_data"
if not os.path.exists(save_path):
os.makedirs(save_path)
graph_data = {"original_graphs": original_graphs,
"augmented_graphs": augmented_graphs,
"gene_labels": gene_labels,
"cell_labels": cell_labels}
save_file = f"{save_path}/{dataset}_cell{cell_numbers}_gene{gene_numbers}_graph{graphs_number}.pkl"
with open(save_file, 'wb') as f:
pickle.dump(graph_data, f)
print(f"Graph data saved to {save_file}")
Graph data saved to ../5_graph_data/merscope_liver_data_region1_portal_cell1713_gene143_graph79172.pkl
step5: Differential gene expression analysis¶
df_merged_region1_central = pd.read_csv("../1.5_benchmark/figure/merfish_liver/df_merged_region1_central.csv")
df_merged_region1_portal = pd.read_csv("../1.5_benchmark/figure/merfish_liver/df_merged_region1_portal.csv")
expr1 = df_merged_region1_central.drop(columns=['cell', 'center_x', 'center_y']).astype(float)
expr2 = df_merged_region1_portal.drop(columns=['cell', 'center_x', 'center_y']).astype(float)
X = pd.concat([expr1, expr2], ignore_index=True)
group = ['central'] * expr1.shape[0] + ['portal'] * expr2.shape[0]
adata = sc.AnnData(X)
adata.obs['group'] = group
adata.var_names = expr1.columns
sc.pp.filter_cells(adata, min_genes=200)
sc.pp.filter_genes(adata, min_cells=10)
sc.pp.normalize_total(adata, target_sum=1e4)
sc.pp.log1p(adata)
sc.tl.rank_genes_groups(adata, groupby='group', method='wilcoxon')
def extract_rank_genes_df(adata, group):
result = adata.uns['rank_genes_groups']
names = result['names'][group]
pvals = result['pvals_adj'][group]
logfc = result['logfoldchanges'][group]
df = pd.DataFrame({'gene': names, 'pvals_adj': pvals, 'log2FC': logfc})
return df
df_deg = extract_rank_genes_df(adata, group='portal')
padj_thresh = 0.05
logfc_thresh = 1
up_genes = df_deg[(df_deg['pvals_adj'] < padj_thresh) & (df_deg['log2FC'] > logfc_thresh)]
down_genes = df_deg[(df_deg['pvals_adj'] < padj_thresh) & (df_deg['log2FC'] < -logfc_thresh)]
print(f"Number of upregulated genes(portal > central): {len(up_genes)}")
print(f"Number of downregulated genes(central > portal): {len(down_genes)}")
up_genes.to_csv("../1.5_benchmark/figure/merfish_liver/upregulated_genes_portal_vs_central.csv", index=False)
down_genes.to_csv("../1.5_benchmark/figure/merfish_liver/downregulated_genes_portal_vs_central.csv", index=False)
up_gene_list = up_genes['gene'].tolist()
down_gene_list = down_genes['gene'].tolist()
gene_list = up_gene_list + down_gene_list
print(f"up_gene_list: {up_gene_list}\n down_gene_list: {down_gene_list}")
if len(up_gene_list) > 0:
print("Plotting upregulated genes heatmap...")
sc.pl.heatmap(adata, var_names=gene_list, groupby='group', cmap='coolwarm', standard_scale='var', dendrogram=False, show=True, save="_up_down_genes")
else:
print("No upregulated genes meet the criteria.")
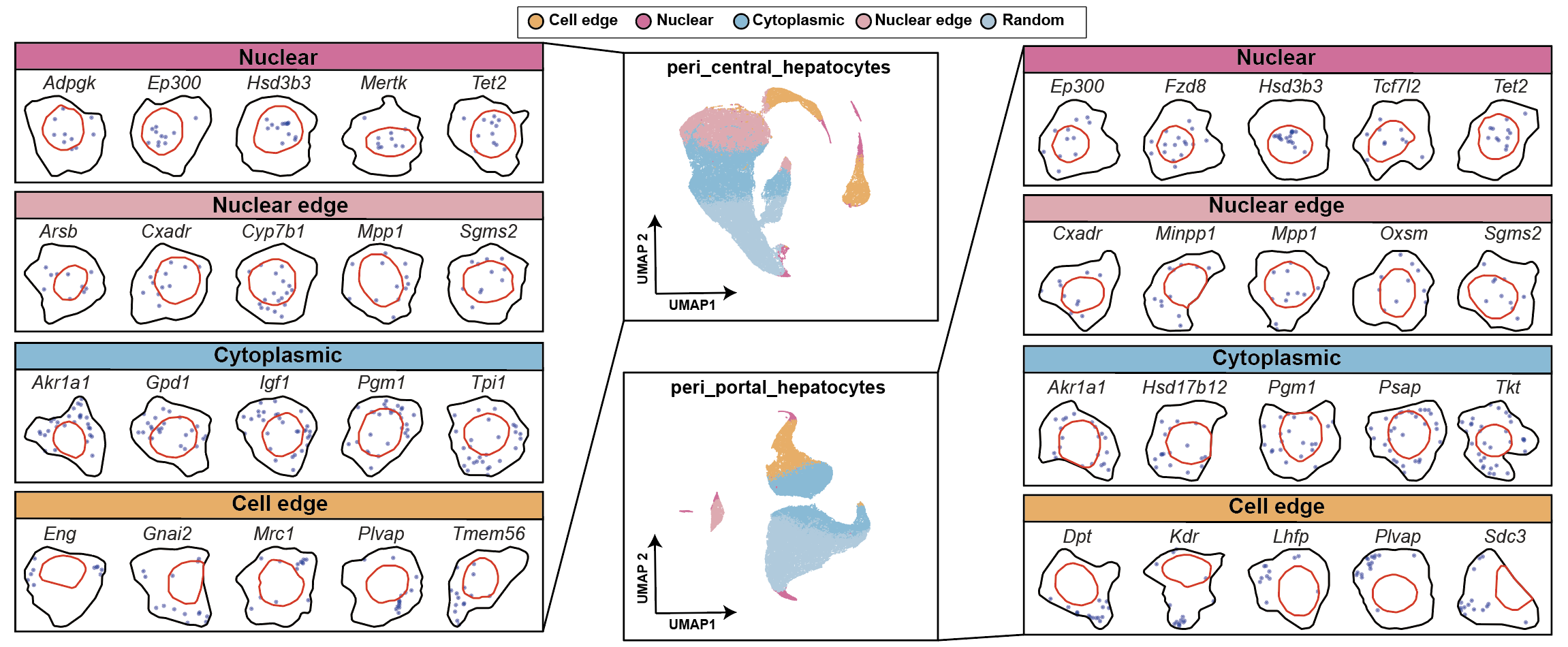
step6: Clustering and identifying spatial localization patterns¶
dataset = "merscope_liver_data_region1_central"
a, b, epoch, lr = 0.2, 0.8, 300, 0.1
df = pd.read_csv(f"../1.5_benchmark/method4_ours/{dataset}/a{a}_b{b}_epoch{epoch}_lr{lr}_pca_ours_df_copy_graph.csv")
df['GRASP'] = df['gmm_clusters5'].replace({0: "Cytoplasmic", 1: "Nuclear edge", 2: "Cell edge", 3: "Random", 4: "Nuclear"})
print(df['GRASP'].value_counts())
color_map = {'Nuclear': '#ed6ca4','Cytoplasmic': '#7bc4e2', 'Protrusion': '#acd372','Nuclear edge': '#fbb05b',
'Cell edge': '#EDABB5', 'Random': '#ACD0E4', 'Foci': '#FFD4AB', 'Radial': '#DDC4E0'}
def plot_tsne_by_label(df, label_col, color_map, title='', save_prefix=None, legend_title='Label', legend_loc='upper left', legend_bbox=(1.1, 1.0), legend_ncol=1, size=10):
plt.figure(figsize=(5, 5))
for label, group in df.groupby(label_col):
plt.scatter(x=group['tsne_x'], y=group['tsne_y'], color=color_map.get(label, '#E9E9E9'), label=label, s=size)
ax = plt.gca()
ax.spines['top'].set_visible(False)
ax.spines['right'].set_visible(False)
ax.spines['left'].set_visible(False)
ax.spines['bottom'].set_visible(False)
plt.xticks([])
plt.yticks([])
plt.grid(False)
plt.title(title)
if save_prefix:
plt.savefig(f'{save_prefix}.png', dpi=300, bbox_inches='tight')
plt.savefig(f'{save_prefix}.pdf', bbox_inches='tight')
plt.savefig(f'{save_prefix}.svg', bbox_inches='tight')
plt.show()
def plot_umap_by_label(df, label_col, color_map, title='', save_prefix=None, legend_title='Label', legend_loc='upper left', legend_bbox=(1.1, 1.0), legend_ncol=1, size=10):
plt.figure(figsize=(5, 5))
for label, group in df.groupby(label_col):
plt.scatter(x=group['umap_x'], y=group['umap_y'], color=color_map.get(label, '#E9E9E9'), label=label, s=size)
ax = plt.gca()
ax.spines['top'].set_visible(False)
ax.spines['right'].set_visible(False)
ax.spines['left'].set_visible(False)
ax.spines['bottom'].set_visible(False)
plt.xticks([])
plt.yticks([])
plt.grid(False)
plt.title(title)
if save_prefix:
plt.savefig(f'{save_prefix}.png', dpi=300, bbox_inches='tight')
plt.savefig(f'{save_prefix}.pdf', bbox_inches='tight')
plt.savefig(f'{save_prefix}.svg', bbox_inches='tight')
plt.show()
plot_tsne_by_label(df=df,label_col='GRASP',color_map=color_map, title='', save_prefix=f'../1.5_benchmark/figure/{dataset}/tsne_ours1',
legend_title='GRASP', legend_loc='lower center', legend_bbox=(0.5, 1.05), legend_ncol=3,size=0.5)
plot_umap_by_label(df=df,label_col='GRASP',color_map=color_map, title='', save_prefix=f'../1.5_benchmark/figure/{dataset}/umap_ours1',
legend_title='GRASP', legend_loc='lower center', legend_bbox=(0.5, 1.05), legend_ncol=3,size=0.5)
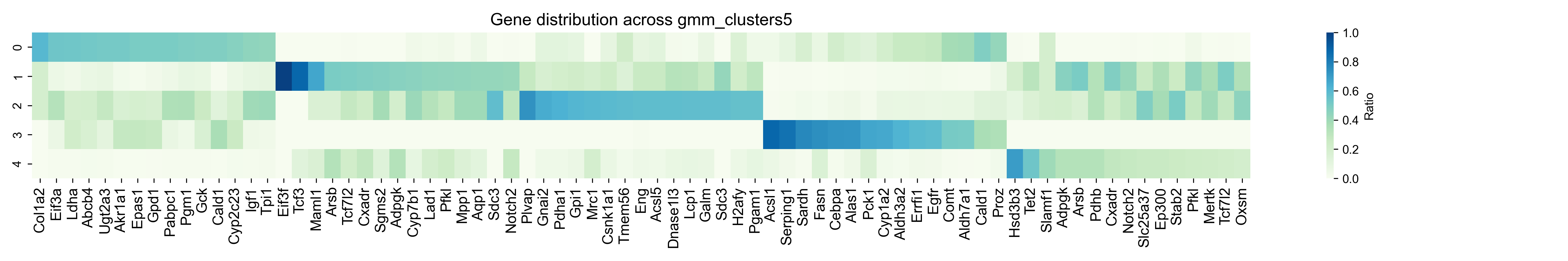
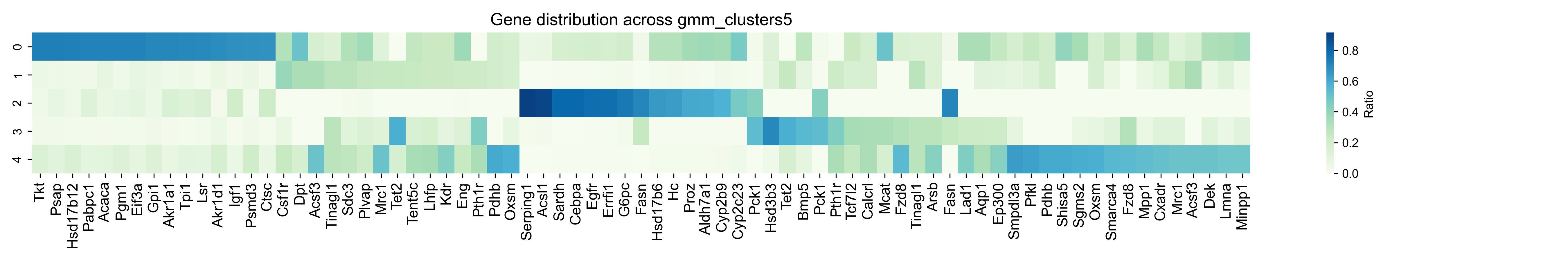
step7: Plot a TSG clustering heatmap¶
dataset = "merscope_liver_data_region1_central"
a, b, epoch, lr = 0.2, 0.8, 300, 0.1
use_pca = True
top_number = 15
method_list = [("km_clusters5", 5), ("gmm_clusters5", 5), ("agg_clusters5", 5)]
output_pdf = f"../1.5_benchmark/figure/{dataset}/a{a}_b{b}_epoch{epoch}_lr{lr}_gene_distribution_heatmaps.pdf"
with PdfPages(output_pdf) as pdf:
for method, n_clusters in method_list:
file_suffix = f"{dataset}/a{a}_b{b}_epoch{epoch}_lr{lr}_{'pca_' if use_pca else ''}ours_df_copy_graph.csv"
df = pd.read_csv(f"../1.5_benchmark/method4_ours/{file_suffix}")
gene_cluster_counts = df.groupby(['gene', method]).size().unstack(fill_value=0)
gene_cluster_counts = gene_cluster_counts[gene_cluster_counts.sum(axis=1) >= 5]
gene_cluster_ratio = gene_cluster_counts.div(gene_cluster_counts.sum(axis=1), axis=0)
top_genes_per_cluster = {
cluster: gene_cluster_ratio[cluster].sort_values(ascending=False).head(100).index.tolist()
for cluster in gene_cluster_ratio.columns
}
top_genes_df = pd.DataFrame({f'Cluster {c}': pd.Series(genes) for c, genes in top_genes_per_cluster.items()})
top_genes_df.to_csv(f"../1.5_benchmark/figure/{dataset}/top_genes_df_{method}_{a}_{b}_{epoch}_{lr}.csv")
genes_of_interest = []
for i in range(n_clusters):
cluster_key = f'Cluster {i}'
if cluster_key in top_genes_df.columns:
genes_of_interest.extend(top_genes_df[cluster_key].dropna().iloc[:top_number].tolist())
df_selected = gene_cluster_ratio.loc[gene_cluster_ratio.index.intersection(genes_of_interest)]
df_selected = df_selected.reindex(genes_of_interest)
plt.figure(figsize=(18, 3))
sns.heatmap(df_selected.T, annot=False, cmap="GnBu", cbar_kws={"label": "Ratio"})
plt.title(f"Gene distribution across {method}", fontsize=14)
plt.xlabel("", fontsize=14)
plt.ylabel("", fontsize=14)
plt.xticks(fontsize=12, rotation=90)
plt.tight_layout()
pdf.savefig()
plt.close()
# plt.show()
print(f"All heatmaps have been saved to :{output_pdf}")
dataset = "merscope_liver_data_region1_central"
output_file = f"../1.5_benchmark/figure/{dataset}/a{a}_b{b}_epoch{epoch}_lr{lr}_matching_results.txt"
all_methods = {
'5_clusters': {
'methods': ['gmm_clusters5', 'km_clusters5', 'agg_clusters5'],
'clusters': ["Cluster 0", "Cluster 1", "Cluster 2", "Cluster 3", "Cluster 4"]
}
}
with open(output_file, 'w', encoding='utf-8') as f:
for setting, conf in all_methods.items():
method_list = conf['methods']
cluster_list = conf['clusters']
for method in method_list:
top_genes_path = f"../1.5_benchmark/figure/{dataset}/top_genes_df_{method}_{a}_{b}_{epoch}_{lr}.csv"
top_genes_df = pd.read_csv(top_genes_path)
f.write("=" * 50 + "\n")
f.write(f"[method:{method}]\n")
for cluster in cluster_list:
element_list = top_genes_df[[cluster]].head(15)[cluster].tolist()
in_df_clusters0 = []
in_df_clusters1 = []
in_df_clusters2 = []
in_df_clusters3 = []
in_df_clusters4 = []
for element in element_list:
if element in df_clusters1:
in_df_clusters0.append(element)
elif element in df_clusters2:
in_df_clusters1.append(element)
elif element in df_clusters3:
in_df_clusters2.append(element)
elif element in df_clusters4:
in_df_clusters3.append(element)
else:
in_df_clusters4.append(element)
f.write(f"\n======= Summary of Results: {method} - {cluster} =======\n")
f.write(f"Number of genes in df_clusters0 (nuclear): {len(in_df_clusters0)} - {in_df_clusters0}\n")
f.write(f"Number of genes in df_clusters1 (nuclear edge): {len(in_df_clusters1)} - {in_df_clusters1}\n")
f.write(f"Number of genes in df_clusters2 (cytoplasmic): {len(in_df_clusters2)} - {in_df_clusters2}\n")
f.write(f"Number of genes in df_clusters3 (cell edge): {len(in_df_clusters3)} - {in_df_clusters3}\n")
f.write(f"Number of genes in df_clusters4 (random): {len(in_df_clusters4)} - {in_df_clusters4}\n")
peri_central_hepatocytes